The Pilar Alcaide Lab

Why Do We Study T Lymphocytes?
T cell recruitment into organs and tissues is a hallmark of several chronic inflammatory processes. Different T cell immune responses are elicited by distinct triggers that are highly context dependent and determine cardiac and vascular pathophysiology in specific disease contexts. We study T lymphocytes because by understanding how these interesting cells modulate other cell functions, we learn new avenues to modulate organ and tissue physiology.
Why Do We Study Heart Failure?
Heart failure (HF) is a leading cause of mortality affecting over 6 million people in the United States. Regardless of the etiology, HF is caused by a decline in left ventricular (LV) function that increases LV pressure and results in LV hypertrophy and fibrosis, a process known as adverse cardiac remodeling. Our lab studies the role of the adaptive immune system in adverse cardiac remodeling. The lab uses well-established experimental mouse models of HF, and a variety of genetically altered mouse models that include gene deletions and gene reporters of T-cell function and allow to investigate the cell specific role of certain molecules in T cell immune responses and in cardiac and vascular remodeling. In collaboration with cardiologists, we apply our mechanistic findings to human disease.

Figure 1: Model of the role of adaptive immunity in non-ischemic HF.
Our lab has demonstrated that in response to high left ventricular pressure (LVP), CD4+ T cells are activated by dendritic cells (DC) specifically in the lymph nodes that drain the heart, are recruited to the heart by interacting with adhesion molecules and chemokines presented by the endothelial cells (EC), where they induce contact dependent cardiac fibrosis upon interactions with cardiac fibroblasts (CFB) and impair cardiac function. We combine in vitro and in vivo studies to understand this process in depth and how specific pathways may be targeted therapeutically. Receptors of gut metabolites involved in HF are also an area of investigation in our lab.
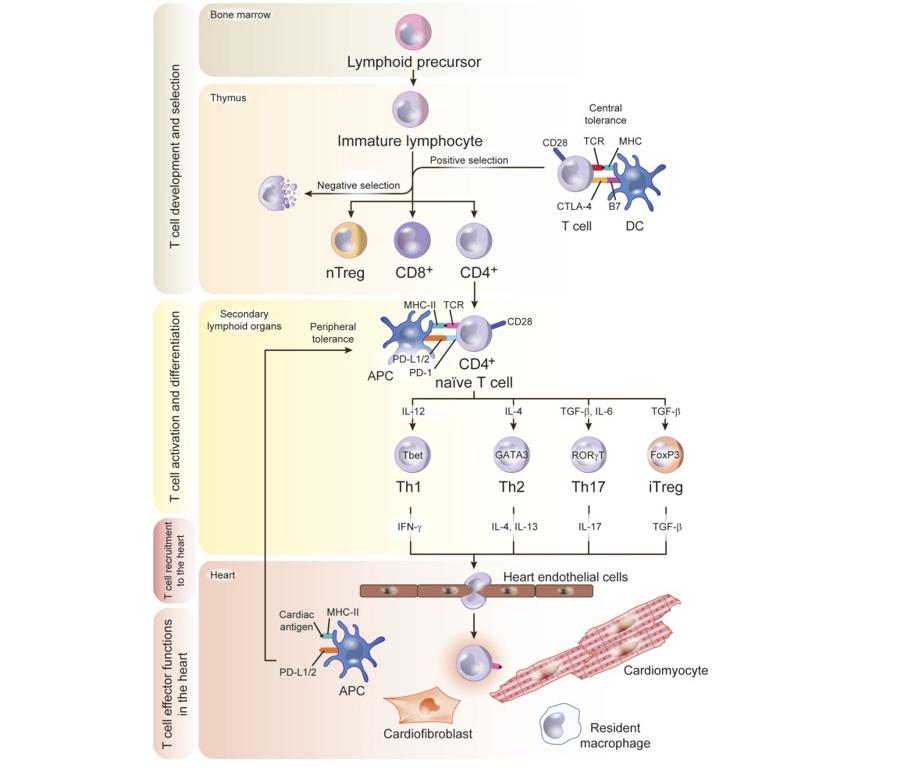
Figure 2. Model of T Cell Development
T-Cell Development, Activation, and Trafficking to the Heart
Lymphoid precursors originate in the bone marrow and traffic to the thymus, where they develop into CD4+ or CD8+ T cells and undergo a complex selection process to discard autoreactive and dysfunctional immature T cells. This crucial step in antigen recognition “learning” is the base of the concept of central tolerance, regulated by signals that involve major histocompatibility complex (MHC) and B7 in the antigen-presenting cell (APC), T-cell receptor (TCR), CD28, and CTLA-4 in the T cell. Positively selected T-cell clones traffic to the secondary lymphoid organs as naïve T cells. If heart inflammation is triggered, dendritic cells (DCs) process cardiac antigens and traffic to secondary lymphoid organs for antigen presentation to naïve T cells that differentiate into effector cell subsets upon antigen recognition. T-cell PD-1 and CD28 and DC PD-L1/2 and B7 signals are an additional mechanism of tolerance in the periphery to antigens that escaped negative selection in the thymus. Effector T cells acquire a migratory profile during differentiation and will be recruited to the heart in response to signals in the heart local environment. Cardiac fibroblasts express the costimulatory molecule CD80 and efficiently capture and process extracellular antigens into small peptides that are uploaded into MHC-II molecules. In response to IFNγ stimulation, cardiac fibroblasts express MHCII and present peptide antigens that induce IFNγ + Th1 cell immune responses and promote cardiac remodeling and dysfunction.
Read more about this work (trainee names in bold font).
Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, Blanton RM, Alcaide P. 2017. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med. Nov 6;214(11):3311-3329.doi: 10.1084/jem.20161791. Abstract
Ngwenyama N, Salvador AM, Velázquez F, Nevers T, Levy A, Aronovitz MJ, Luster AD, Huggins GS, Alcaide P. 2019. CXCR3 regulates CD4+ T cell cardiotropism in pressure overload induced cardiac dysfunction. JCI Insight 4 (7) pii: 125527. doi: 10.1172/jci.insight.125527. Abstract
Blanton RM, Carrillo-Salinas FJ, Alcaide P. 2019. T-cell recruitment to the heart: friendly guests or unwelcome visitors? Am J Physiol Heart Circ Physiol. Jul 1;317(1):H124-H140. Abstract
Ngwenyama N, Kirabo A, Aronovitz M, Velázquez F, Carrillo-Salinas F, Salvador AM, Nevers T, Amarnath V, Tai A, Blanton RM, Harrison DG, Alcaide P. 2021. Isolevuglandin-modified cardiac proteins drive CD4+ T cell activation in the heart and promote cardiac dysfunction. Circulation. doi: 10.1161/CIRCULATIONAHA.120.051889. Abstract
Ngwenyama N, Kaur K, Bugg D, Theall B, Aronovitz M, Berland R, Panagiotidou S, Genco C, Perrin MA, Davis J, Alcaide P. 2022. Antigen presentation by cardiac fibroblasts promotes cardiac dysfunction. Nat Cardiovasc Res. Aug;1(8):761-774. doi: 10.1038/s44161-022-00116-7. Abstract
Ongoing Projects in the Alcaide Lab
Cardiac T Cell Response in Heart Failure
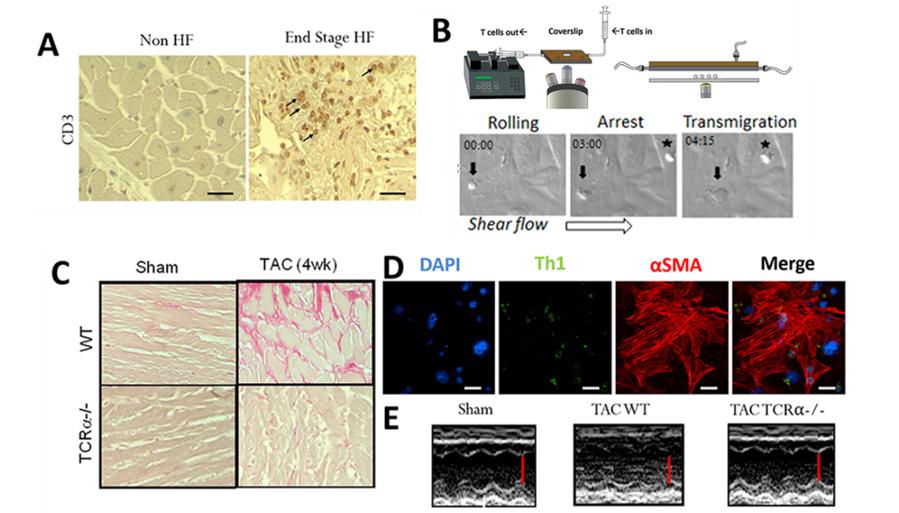
Figure 3. Cardiac T cell infiltration and T cell cross talk with cardiac endothelial cells and fibroblasts. A. T cells are found infiltrated in the human failing heart. Immunohistochemistry for CD3+ T cells in human heart ventricular tissue. Arrows indicate representative T cells in the heart of a HF patient who underwent Left ventricular assisted device (LVAD) placement. Control heart was obtained from NDRI. B. T cells activated in response to TAC adhere and transmigrate across mouse heart endothelial cells. Example of CD4+ T cells isolated from cardiac draining lymph nodes of mice 4 weeks after TAC adhering (indicated with a *) and transmigrating (indicated with an arrow) across 4h TNFα activated mouse heart endothelial cells under shear flow conditions in vitro. C. T cell deficient mice (TCRα-/-) do not develop cardiac fibrosis in response to TAC. Picrosirius red staining of heart sections from Sham and TAC WT and TCRα-/- mice indicating absence of collagen deposition (in pink) in the absence of T cells. D. T cell contact dependent cardiac fibrosis. T cell (green) contact with cardiac fibroblasts induces αSMA expression (shown in red) and transformation to pro-fibrotic myofibroblasts. E. TCRα-/- are protected from TAC induced HF. M mode Echocardiography images show preserved systolic function in TCRα-/- mice in response to TAC as compared to WT TAC mice. Scale bar is indicated in red.
Read more about this work (trainee names in bold font).
Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, Blanton RM, Alcaide P. 2017. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med. Nov 6;214(11):3311-3329.doi: 10.1084/jem.20161791. Abstract
Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velázquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P. 2015. Left ventricular T-cell recruitment contributes to the pathogenesis of heart failure. Circ Heart Fail. Jul;8(4):776-87.doi: 10.1161/CIRCHEARTFAILURE.115.002225. Abstract
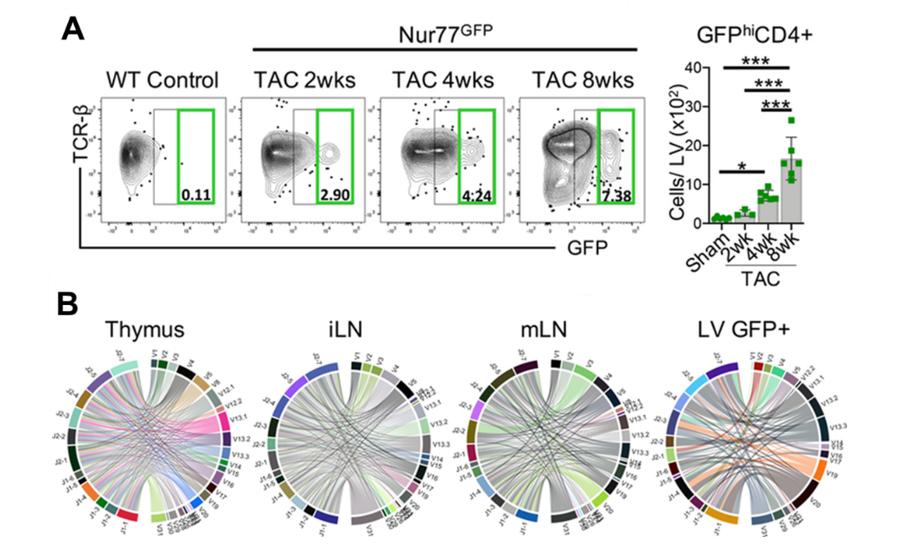
Figure 4. CD4+ T cells are activated via the TCR in the LV in response to TAC. A. Nur77GFP mice, reporters of TCR engagement, underwent Sham surgery for 8wks and TAC surgery for 2–8wks. Leukocytes were isolated from the LV and the total number of GFPhiCD4+ T cells was quantified by FACS. B. Bulk TCR clonal analysis after 8wks of TAC surgery (n=3 TAC Nur77GFP mice pooled) was performed on 5000 CD4+ T cells sorted from the thymus, inguinal lymph nodes (iLN), mediastinal lymph nodes (mLN) and 5000 GFP+CD4+ T cells sorted from the LV by next generation RNA sequencing. The frequency of the most abundant 20 clones was enriched in the LV and to some extent, in the mLNs, whereas the thymus and iLN, further from the heart, were not enriched for unique CD4+ T cells clones and showed the expected enhanced clonal diversity. CD4+ T cell clonotypes are represented in chord diagrams, with ribbons proportionately scaled by V-J pair frequency connecting segment pairs. The thickness proportionally scales to the relative abundance of clones using the V-J segment pairs.
Read more about this work (trainee names in bold font).
Ngwenyama N, Kirabo A, Aronovitz M, Velázquez F, Carrillo-Salinas F, Salvador AM, Nevers T, Amarnath V, Tai A, Blanton RM, Harrison DG, Alcaide P. 2021. Isolevuglandin-modified cardiac proteins drive CD4+ T cell activation in the heart and promote cardiac dysfunction. Circulation. doi: 10.1161/CIRCULATIONAHA.120.051889. Abstract
Fibroblast - T Cell Cross Talk - Stromal Immunology in Cardiac Pathophysiology
Cardiac fibrosis (CF) is a hallmark of HF causing decreased mechanical function of the heart. Cardiac Fibroblasts (CFBs), the main drivers of CF, are activated in HF by numerous stimuli including signals from immune cells. While the extent of CF is linked to worse prognosis in HF patients, the lack of clinically available antifibrotic therapies highlights a need for greater understanding of the fibrotic mechanisms of HF. Our lab has shown that T-cell deficient mice do not develop CF, and that T cell interactions with CFB are needed for CFB transformation. During this interaction, we have identified a two-way crosstalk in which T cells induce CFB transformation, and CFB, in turn, function as non-professional antigen presenting cells that induce T cell activation. Specific cardiac fibroblast deletion of the major histocompatibility complex II (MHC-II), required for antigen presentation, prevents cardiac fibrosis induced by TAC.
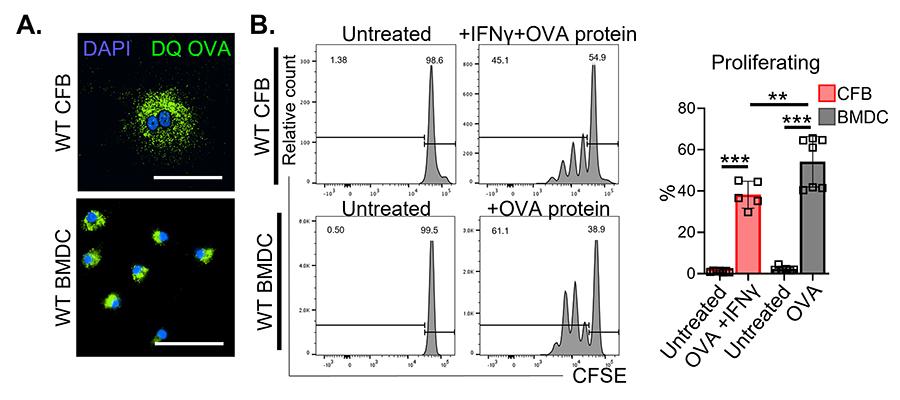
Figure 5. Cardiac fibroblasts take up, process and present antigens to CD4+ T cells in vitro. A. Immunofluorescence microscopy in cardiac fibroblasts and dendritic cells (DCs) to evaluate uptake and intracellular processing of fluorescent ovalbumin protein (DQ OVA). B. WT cardiac fibroblasts and DCs were treated with purified OVA protein (100 μg ml−1) overnight and co-cultured with CFSE-labeled naïve OTII CD4+ T cells in the presence of IFN-γ (100 U ml−1) for 72 h to evaluate CD4+ T cell proliferation by flow cytometry.
Read more about this work (trainee names in bold font).
Ngwenyama N, Kaur K, Bugg D, Theall B, Aronovitz M, Berland R, Panagiotidou S, Genco C, Perrin MA, Davis J, Alcaide P. 2022. Antigen presentation by cardiac fibroblasts promotes cardiac dysfunction. Nat Cardiovasc Res. Aug;1(8):761-774. doi: 10.1038/s44161-022-00116-7. Abstract
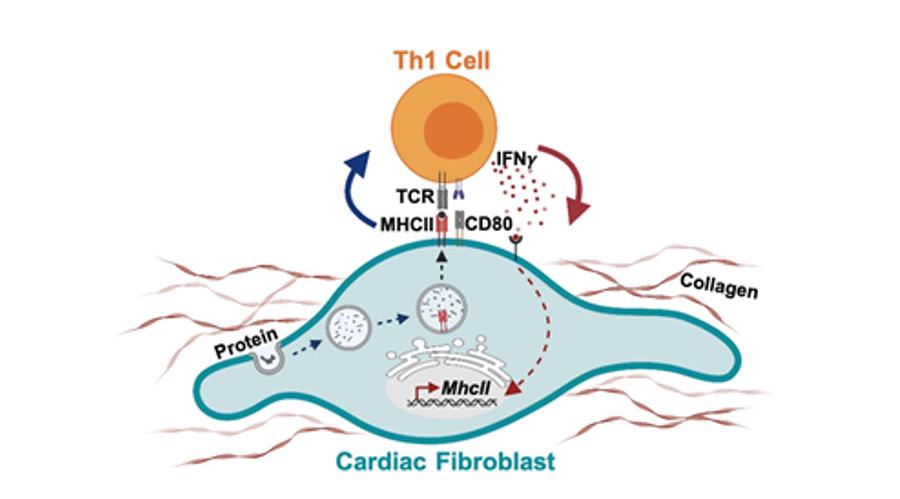
Figure 6. Bidirectionality of cardiac fibroblast and Th1 cell interactions. Cardiac fibroblasts express the costimulatory molecule CD80 and efficiently capture and process extracellular antigens into small peptides that are uploaded into MHC-II molecules. In response to IFNγ stimulation, cardiac fibroblasts express MHCII and present peptide antigens that induce IFNγ + Th1 cell immune responses and promote cardiac remodeling and dysfunction.
Read more about this work (trainee names in bold font).
Ngwenyama N, Kaur K, Bugg D, Theall B, Aronovitz M, Berland R, Panagiotidou S, Genco C, Perrin MA, Davis J, Alcaide P. 2022. Antigen presentation by cardiac fibroblasts promotes cardiac dysfunction. Nat Cardiovasc Res. Aug;1(8):761-774. doi: 10.1038/s44161-022-00116-7. Abstract
Ongoing studies in the Alcaide lab are focused on understanding the newly appreciated role of CFBs as non-professional antigen presenting cells in the context of heart failure. We are investigating the regulation ofMHC-II in CFB and its role in cardiac fibroblast states. Our lab is also investigating how the Aryl Hydrocarbon Receptor (AhR), a widely expressed transcription factor involved in responding to metabolic and environmental cues, functions in modulating CFBs as antigen presenting cells and in CFB transformation.
Novel Regulators of Leukocyte Trans-endothelial Migration (TEM) and Their Contribution to Cardiac and Vascular Pathophysiology
The vascular endothelium plays a central role in modulating T cells extravasation to tissues during the immune response. We found that the Stimulator of Interferon genes (STING), a key modulator of anti-viral responses in immune cells, is expressed in endothelial cells and contributes to T cell transendothelial migration induced by TNFα in vitro and in vivo.
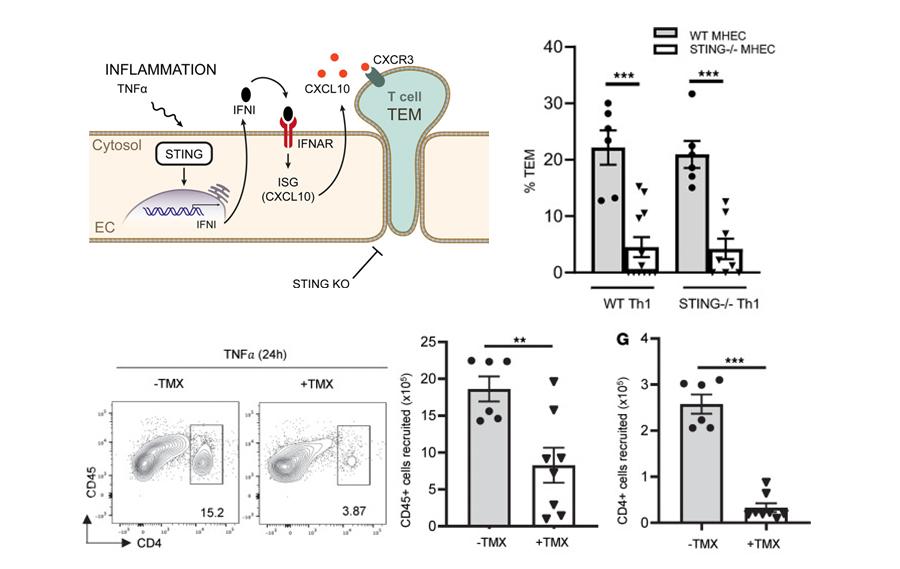
Figure 7. Endothelial Cell STING contributes to TNF-α– induced T cell TEM and T cell recruitment to sites of inflammation. A. Schematic representation indicating that the absence of endothelial STING impairs TEM in a IFN-I dependent manner resulting in impaired CXCL10 necessary for T cell TEM. B. STING deficiency in mouse heart endothelial cells (MHEC) but not in T cells results in decreased TEM under flow conditions in in response to TNF-α in vitro. C-D. Decreased TNF-α induced T cell recruitment into the peritoneal cavity of EC-STING–/– mice (Cad5ERT2Stingfl/fl treated with tamoxifen (TMX). C. Representative flow cytometric panels, D. Quantification of total CD45+ and CD4+ cells recruited cells to the peritoneal cavity 24 h after TNF-α injection.
Read more about this work (trainee names in bold font).
Anastasiou M, Newton GA, Kaur K, Carrillo-Salinas FJ, Smolgovsky SA, Bayer AL, Ilyukha V, Sharma S, Poltorak A, Luscinskas FW, Alcaide P. 2021. Endothelial STING controls T-cell transmigration in an IFN-I dependent manner. JCI Insight. 22:149346. doi: 10.1172/jci.insight.149346. Abstract
Ongoing projects in the lab are investigating the role of endothelial STING in adverse cardiac remodeling using experimental models of HF, as well as in vascular remodeling using experimental models of endothelial injury.
T Cell Immune Responses in Heart Failure with Preserved Ejection Fraction (HFpEF) and Other HF Etiologies
Heart Failure with Preserved Ejection Fraction (HFpEF) is a condition which comprises half of HF diagnoses in which cardiac relaxation is impaired despite normal contractile function. There are few direct treatment options for this condition that is rising in incidence, and for which none of the current HF therapies work. A key characteristic of HFpEF is systemic chronic low-grade inflammation. The lab uses a pre-clinical mouse model of cardiometabolic HFpEF induced by obesity and hypertension, two of the most prevalent HFpEF comorbidities, to investigate the T cell signatures imprinted by the combination of these two co-morbidities.
Additionally, there are ongoing projects in the lab focusing on cardio-immuno-oncology, aiming to understand novel mechanisms of immune activation in melanoma and in chemotherapy induced cardiotoxicity. The ultimate goal is to understand new ways to alter T cell function that can be used for anti-tumor immunity while decreasing cardiotoxicity by dampening T cell function.
Mentoring and Research Highlights
Everyone in the Alcaide lab benefits from continuous learning and peer mentoring. Here are some reading materials about research and mentoring from members of the Alcaide lab.
Read about the Alcaide lab in Tufts Now.
See the Alcaide lab in GSBS News.